This piece originally appeared in the Timmerman Report.
What do the polio virus, baseball pitch choice and cancer have in common?
The answer, of course, is sequencing. But not in the “figure out the DNA” way (although that’s involved). Instead in the “what comes first” way. Confused? Read on!
A big perk of Seattle is proximity to great institutions of biomedical research like the University of Washington and the Fred Hutchinson Cancer Research Center. Ever since my graduate student days in genetics at UC-Berkeley I’ve enjoyed going to seminars–especially seminars that are outside my field of study. Very little beats a good seminar for giving you a quick, condensed view of the state of a field of research. A bad seminar…well…we all could use more sleep, right?
In early October, Raul Andino of UCSF came to the Hutch to talk about his work on viral evolution. His team has been examining a clever real-world system to track the evolution of viruses. The near-eradication of polio (one of the great public heath victories of the past century) has led to the curious problem that as of the middle of this year most new cases of polio arose as a result of vaccination efforts. The live, attenuated vaccine that’s used in the developing world can, in very rare cases, mutate in just the wrong ways in its host, leading to the creation of a virulent strain that can infect others. In the US we use an inactivated polio vaccine which requires several injections; in much of the developing world the oral polio virus is preferred due to its ease of administration, lower cost, and immunization profile. The Andino lab realized that by studying these isolated outbreaks, which all originated with the same, genetically identical progenitor, they could test a hypothesis about the adaptive landscape of virulence evolution.
Are there many paths to virulence or just a few? Are different phenomenon fated to be in some way or are there many alternative paths? If these escaping strains appeared different from one another at the sequence level, that would suggest multiple paths to virulence. If phylogenetic analysis revealed commonalities, that would suggest the adaptive landscape is constrained.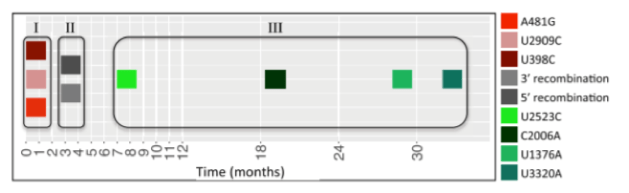
This figure from the Andino Lab paper shows that, based on both observational and experimental data, the second case holds. In most of the reverting strains many of the same important mutations were seen across strains, likely arising via parallel selection. The boxed area labeled “I” represents key mutations that happen first and act to reverse some of the initial genetic elements that make the vaccine attenuated in the first place. Once the strain becomes more able to replicate, recombination events with human enteroviruses of species C (HEV-C) provide the polio virus with presumed more favorable genes from the HEV-C (box “II”). The last set of mutations, in the box labeled “III” are less understood, but common enough to suggest a further contribution to fitness.
It surprised me that there’s a definite sequence of events. “A” must precede “B,” which in turn must happen before “C”. Their visualization of the adaptive pathway leading up to the peak of virulence shows this linear path (see below). I often think of evolutionary selection as exploring the near-infinite sandbox of possibility. And in one sense it does. Most vaccinations never lead to a virulent strain; the mutations that occur explore the evolutionary space but also peter out, never to be analyzed. But to reach the apex of virulence? There appears to be one main path, with the caveat that the starting point was identical. Still to see so many independent virulence events across several different human populations recapitulate the same path is pretty strong evidence.
So in this case the sequencing of events matters. Which brings me to baseball.
An active area of research in baseball is pitch sequencing: what effect does the order and location of pitches have on the outcome of an at-bat? Statisticians are hard at work. But don’t take my word for it! Here’s the lede from a 2015 article by Peter Bonney in the Hardball Times:

The null hypothesis would be that order and location of pitches don’t matter. Few if any believe this but teasing out the effect is, as Bonney states, difficult to do. That article goes through some of the challenges, most of which involve the inherent variability of context in baseball. Pitches aren’t context-neutral. A pitch thrown at the start of an at-bat is not the same as one thrown with the count 0-2 or 1-1. And what about number of outs in the inning? Men on base? Current score and inning? Complexity complicates analysis: try to limit your analysis to specific contextual elements and you quickly shrink the sample size. Pitchers in Major League Baseball (MLB) throw over 700,000 pitches during a typical season, but that’s not really all that many, especially for events (hits, strikeouts, etc) that have a large random component and where the expected effect sizes may be quite small.
However, just because a problem is hard doesn’t mean people won’t try to figure it out. This analysis from 2014 approached the problem from the standpoint of sequential pitches and the batter’s eye. The author, Jon Roegele, started with the knowledge that most batters must decide whether to swing or not before the pitch has traveled much further than 33 feet from the pitcher. A 90 mph pitch takes just 0.25 seconds to travel that distance. That’s not much time for the batter to react. Imagine what that’s like? Now let’s say you’re facing Los Angeles Dodgers star pitcher Yu Darvish. His pitches move like this (You really need to click on that and mouse over the gif to watch. It’s amazing!). Not easy.
Given this, Roegele hypothesized that pitches that looked most similar to each other up to that 33 foot point while crossing the plate at different locations would lead to favorable outcomes for the pitcher—the batter is fooled. His initial analysis of every two-pitch combination over the 2014 seasons looks like this:

His output metric was swinging strike rate: how often the batter swings and misses at the second pitch. Let’s just look at the top row. Those are pitches that, at 33 feet from the plate, appeared within an inch of the location of the previous pitch. As you go right along the top row the final distance between pitch locations as the pitch crosses home plate increases. Green represents an increase in swinging strike rate and the black lines highlight the sector of pitch combinations that appear to increase the likelihood of a swing and a miss. As hypothesized, if the second pitch seems to be traveling along the same path as the previous one, but ends up in a different location, the batter is more likely to swing and miss.
This doesn’t take into account any of the factors that Peter Bonney pointed out—all those in-game context factors, but to see a pattern is encouraging. It’s one of many approaches that are teasing out cause and effect and probabilities in pitch sequencing in baseball, showing that sequencing of events matters.
Which brings me to cancer.
Solving viral evolution or pitch sequencing, while challenging is, in my opinion, dwarfed in difficulty by the complexity and challenge in answering the analogous question in cancer. What is the effect of the sequence of mutations that give rise to a malignancy? Does the order of accumulation of driver mutations matter? Does it matter which pathways get mutated first? Or is “Tumor Mutational Burden” the sheer number of mutations in a given tumor, what really matters?
The biological and therapeutic implications are clear. Unveiling the mutational burden across a wide spectrum of cancers has been one of the greatest applications of next-generation sequencing technologies and has led to more precise treatment strategies. We now know cancers, in some cases, can be classified and treated based on specific driver mutations agnostic of tissue of origin. Still, many of these studies have been done in a cross-sectional way and also late in tumor development. Almost by default studied tumors were large enough to be detected and resected. What we need, ideally, is an understanding of how cancers progress from an early stage and whether, like polio, there are sequential constraints on the incidence of mutations. And whether, like pitch sequencing, specific temporal combinations change the probability of outcomes.
These aren’t easy studies to do, not least because longitudinal sequencing of a patient’s tumor burden isn’t anywhere close to standard practice. However, cancer researchers are performing studies in this area—one seminal study from the University of British Columbia demonstrated the utility of repeatedly sequencing a metastatic tongue cancer over time to guide treatment. There are several fascinating approaches researchers are taking from angles such as epidemiology and computer modeling to derive parameters such as how many mutations are likely needed to reach tumorigenicity.
But most of these studies take place long after a cancer has grown and mutated for a while. We can’t know which mutation came first. Animal model systems don’t seem like the answer either, as this recent paper shows. Taking patient tumors and transplanting them into mice as xenografts led to mouse-specific mutational patterns. While we all love mice, we aren’t really interested in curing mouse cancer. However, as has happened so often over the past twenty years, sequencing may come to the rescue. If liquid biopsy companies like Grail are successful in detecting circulating tumor cells before there is any detectable sign of a solid tumor in the body, these cells will represent the earliest look yet at the etiology of cancer. Kind of like the telescopes that keep pushing back our view of the birth of the universe.
If this look shows very specific pathways that seem highly represented in many types of nascent tumors, you’d best get out of the way as biopharma companies charge en masse toward that set of targets for cancer prevention. Talk about the ultimate in lucrative maintenance therapy. And not far behind would come the CRISPR-wielding scientists seeking to engineer mutational robustness and additional redundancy into these pathways, with the long-term goal of trying to cancer-proof future generations of humanity. Although, as I heard at a recent symposium on genome editing with CRISPR, we need to tread lightly in thinking any alterations we make in the genome will be restricted with no unintended consequences.
Solving sequencing of events in cancer may turn out to be the killer app for liquid biopsies. I’m less confident in them as a cancer detection tool (I fear false positives and lack of treatments will make the field unprofitable). In the meantime, I’ll keep pondering the deep question Ebenezer asked of the Ghost of Christmas Yet To Be: “Are these the shadows of the things that Will be, or are they shadows of the things that May be only?”
